This function computes Linearised Methylated Haplotype Load (\(lMHL\)) per genomic position.
Usage
generateMhlReport(
bam,
report.file = NULL,
cytosine.context = c("CG", "CHG", "CHH", "CxG", "CX"),
max.haplotype.window = 0,
filter.reads = TRUE,
min.haplotype.length = 0,
max.outofcontext.beta = 0.1,
...,
gzip = FALSE,
verbose = TRUE
)Arguments
- bam
BAM file location string OR preprocessed output of
preprocessBamfunction. Read more about BAM file requirements and BAM preprocessing atpreprocessBam.- report.file
file location string to write the \(lMHL\) report. If NULL (the default) then report is returned as a
data.tableobject.- cytosine.context
string for a cytosine context that defines a haplotype:
"CG" (the default) – CpG cytosines only (called as zZ)
"CHG" – CHG cytosines only (xX)
"CHH" – CHH cytosines only (hH)
"CxG" – CG and CHG cytosines (zZxX)
"CX" – all cytosines; this, as well as the other non-CG contexts, may have little sense but still included for consistency
If \(lMHL\) calculations are needed for all three possible cytosine contexts independently, one has to run this function for each required `cytosine.context` separately, because `cytosine.context`=="CX" assumes that any cytosine context is allowed within the same haplotype. This behaviour may change in the future.
- max.haplotype.window
non-negative integer for maximum value of \(L'\) in \(lMHL\) formula. When 0 (the default), calculations are performed for the full haplotype length (\(L'=L\), although the maximum value is currently limited to 65535). Having no length restrictions make sense for short-read sequencing when the length of the read is comparable to the length of a typical methylated block, the depth of coverage is high, and the lengths of all reads are roughly equal. However, calculations using non-restricted haplotype length are meaningless for long-read sequencing — when the same read may cover a number of regions with very different methylation properties, and reads themselves can be of a very different length. In the latter case it is advised to limit the `max.haplotype.window` to a number of cytosines in a typical hypermethylated region. For thorough explanation and more examples, see Details section and vignette.
- filter.reads
boolean defining if sequence reads with too high out-of-context cytosine methylation (specified by `max.outofcontext.beta`) or too few within-the-context bases (specified by `min.haplotype.length`) should be filtered out. Default: TRUE. Filtering is strongly recommended for short-read sequencing (bisulfite or enzymatic) because it removes reads from incompletely converted DNA molecules.
- min.haplotype.length
non-negative integer for minimum length of a haplotype (default: 0 will include haplotypes of any length). When `min.haplotype.length`>0, reads (read pairs) with fewer than `min.haplotype.length` cytosines within the `cytosine.context` are skipped. This option has no effect when read filtering is disabled.
- max.outofcontext.beta
real number in the range [0;1] (default: 0.1). Reads (read pairs) with average beta value for out-of-context cytosines above this threshold (e.g., reads resulting from incompletely bisulfite-converted templates) are skipped. Value of 1 disables filtering by out-of-context methylation. This option has no effect when read filtering is disabled.
- ...
other parameters to pass to the
preprocessBamfunction. Options have no effect if preprocessed BAM data was supplied as an input.- gzip
boolean to compress the report (default: FALSE).
- verbose
boolean to report progress and timings (default: TRUE).
Value
data.table object containing \(lMHL\)
report or NULL if report.file was specified. The
report columns are:
rname – reference sequence name (as in BAM)
strand – strand
pos – cytosine position
context – methylation context
coverage – number of reads (read pairs) that include this position
length – average length of a haplotype, i.e., average number of cytosines within `cytosine.context` for reads (read pairs) that include this position
lmhl – \(lMHL\) value
Details
The function reports Linearised Methylated Haplotype Load (\(lMHL\)) at the level of individual cytosines using BAM file location or preprocessed data as an input. Function uses the following formula:
$$lMHL=\frac{\sum_{i=1}^{L'} w_{i} \times MH_{i}}{\sum_{i=1}^{L'} w_{i} \times H_{i}}$$
where \(L'\) is the length of a calculation window (e.g., number of CpGs; \(L' \le L\), where \(L\) is the length of a haplotype covering current genomic position), \(MH_{i}\) is a number of fully successive methylated stretches with \(i\) loci within a methylated stretch that overlaps current genomic position, \(H_{i}\) is a number of fully successive stretches with \(i\) loci, \(w_{i}\) is a weight for \(i\)-locus haplotype (\(w_{i}=i\)).
This formula is a modification of the original Methylated Haplotype Load (MHL) formula that was first described by Guo et al., 2017 (doi: 10.1038/ng.3805):
$$MHL=\frac{\sum_{i=1}^{L} w_{i} \times \frac{MH_{i}}{H_{i}}}{\sum_{i=1}^{L} w_{i}}$$
where \(L\) is the length of a longest haplotype covering current genomic position, \(\frac{MH_{i}}{H_{i}}=P(MH_{i})\) is the fraction of fully successive methylated stretches with \(i\) loci, \(w_{i}\) is a weight for \(i\)-locus haplotype (\(w_{i}=i\)).
The modifications to original formula are made in order to:
provide granularity of values — the original MHL formula gives the same MHL value for every cytosine of a partially methylated haplotype (e.g., MHL=0.358 for each cytosine within a read with methylation call string "zZZZ"). In contrast, \(lMHL\)==0 for the non-methylated cytosines (e.g., \(lMHL\)==c(0, 0.5, 0.5, 0.5) for cytosines within a read with methylation call string "zZZZ").
enable calculations for long-read sequencing alignments — \(lMHL\) calculation window can be limited to a particular number of cytosines. This allows to use the formula for very long haplotypes as well as to compare values for sequencing data of varying read length.
reduce the complexity of MHL calculation for data of high breadth and depth — \(lMHL\) values for all genomic positions can be calculated using a single pass (cycling through reads just once) as the linearised calculations of numerator and denominator for \(lMHL\) do not require prior knowledge on how many reads cover a particular position. This is achieved by moving \(H_{i}\) multiplier to the denominator of the \(lMHL\) formula.
These modifications make \(lMHL\) calculation similar though non-equivalent to the original MHL. However, the most important property of MHL — emphasis on hypermethylated blocks — is retained. And in return, \(lMHL\) gets better applicability for analysis of sequencing data of varying depth and read length.
Other notes on function's behaviour:
Methylation string bases in unknown context ("uU") are simply ignored, which, to the best of our knowledge, is consistent with the behaviour of other tools.
Cytosine context present in more than 50% of the reads is assumed to be correct, while all bases at the same position but having other methylation context are simply ignored. This allows reports to be prepared without using the reference genome sequence.
See also
`values` vignette for a comparison and visualisation of epialleleR output values for various input files. `epialleleR` vignette for the description of usage and sample data.
preprocessBam for preloading BAM data,
generateCytosineReport for other methylation statistics at the
level of individual cytosines,
generateBedReport for genomic region-based statistics,
generateVcfReport for evaluating epiallele-SNV associations,
extractPatterns for exploring methylation patterns
and plotPatterns for pretty plotting of its output,
generateBedEcdf for analysing the distribution of per-read
beta values.
Examples
capture.bam <- system.file("extdata", "capture.bam", package="epialleleR")
# lMHL report
mhl.report <- generateMhlReport(capture.bam)
#> Checking BAM file:
#> short-read, paired-end, name-sorted alignment detected
#> Reading paired-end BAM file
#> [0.013s]
#> Preparing lMHL report
#> [0.019s]
# lMHL report with a `max.haplotype.window` of 1 is identical to a
# conventional cytosine report (or nearly identical when sequencing errors
# are present)
mhl.report <- generateMhlReport(capture.bam, max.haplotype.window=1)
#> Checking BAM file:
#> short-read, paired-end, name-sorted alignment detected
#> Reading paired-end BAM file
#> [0.012s]
#> Preparing lMHL report
#> [0.018s]
cg.report <- generateCytosineReport(capture.bam, threshold.reads=FALSE)
#> Checking BAM file:
#> short-read, paired-end, name-sorted alignment detected
#> Reading paired-end BAM file
#> [0.012s]
#> Filtering reads
#> [0.001s]
#> Preparing cytosine report
#> [0.010s]
identical(
mhl.report[, .(rname, strand, pos, context, value=lmhl)],
cg.report[ , .(rname, strand, pos, context, value=meth/(meth+unmeth))]
)
#> [1] TRUE
# Long-read sequencing with filtering disabled, using window of 10 CpGs
long.bam <- system.file("extdata", "longread.bam", package="epialleleR")
long.data <- preprocessBam(bam=long.bam, min.mapq=30, min.baseq=20,
min.prob=178)
#> Checking BAM file:
#> long-read, single-end, unsorted alignment detected
#> Reading single-end BAM file
#> [0.006s]
mhl.report <- generateMhlReport(bam=long.data, max.haplotype.window=10,
filter.reads=FALSE)
#> Preparing lMHL report
#> [0.051s]
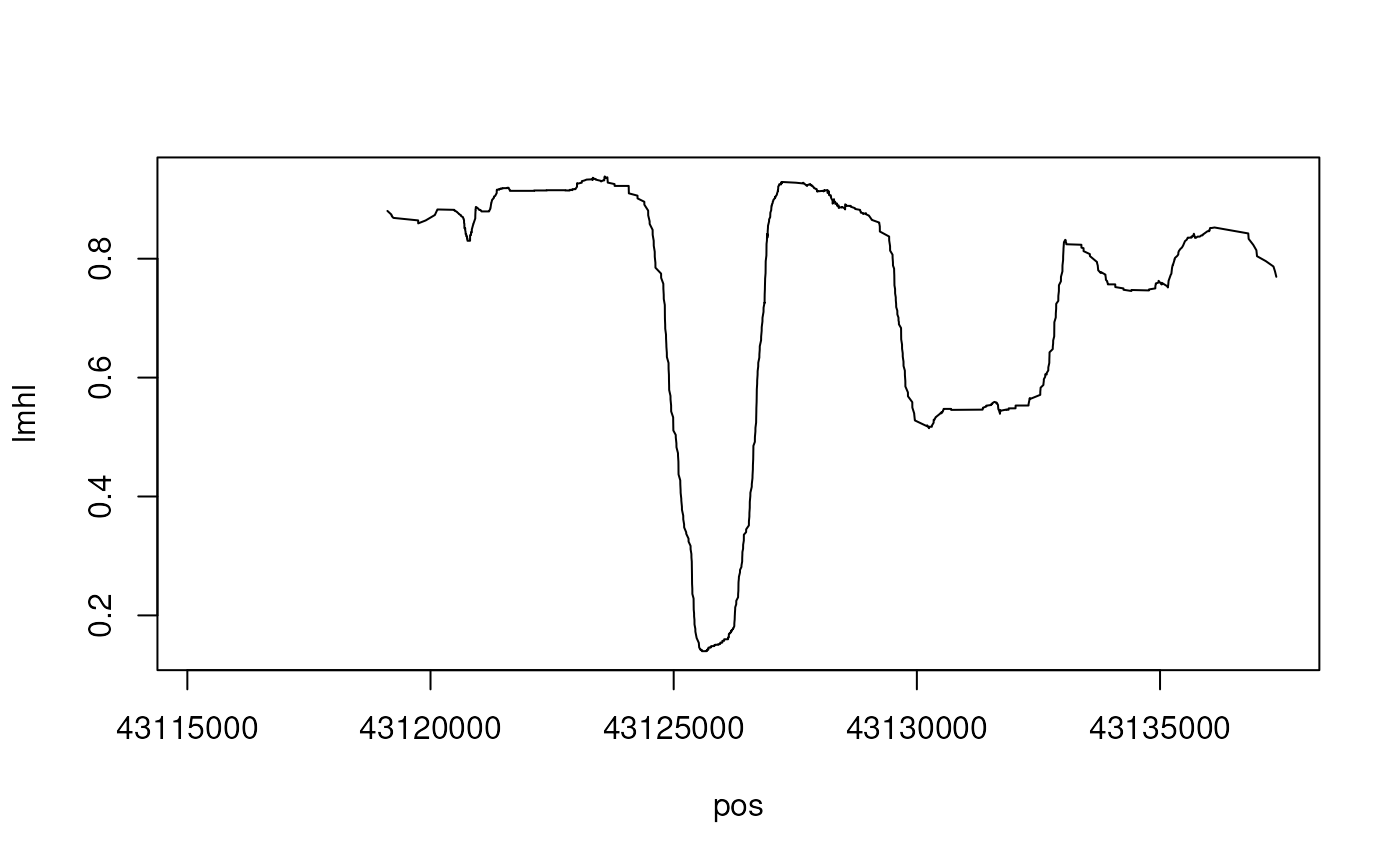
plot(mhl.report[, .(pos, lmhl=data.table::frollmean(lmhl, 100))], type="l")
 ## toy examples to illustrate the logic of computations
temp.bam <- tempfile(fileext=".bam")
# case 1: fully methylated haplotype
simulateBam(output.bam.file=temp.bam, rname="chr1", XG="CT",
XM="h..Z..Z.Z..Z...Z.h.")
#> Writing sample BAM
#> [0.003s]
#> [1] 1
generateMhlReport(temp.bam)
#> Checking BAM file:
#> short-read, single-end, unsorted alignment detected
#> Reading single-end BAM file
#> [0.001s]
#> Preparing lMHL report
#> [0.001s]
#> rname strand pos context coverage length lmhl
#> <fctr> <fctr> <int> <fctr> <int> <num> <num>
#> 1: chr1 + 4 CG 1 5 1
#> 2: chr1 + 7 CG 1 5 1
#> 3: chr1 + 9 CG 1 5 1
#> 4: chr1 + 12 CG 1 5 1
#> 5: chr1 + 16 CG 1 5 1
# case 2: incompletely methylated haplotype
simulateBam(output.bam.file=temp.bam, rname="chr1", XG="CT",
XM="h..Z..Z.z..Z...Z.h.")
#> Writing sample BAM
#> [0.003s]
#> [1] 1
generateMhlReport(temp.bam)
#> Checking BAM file:
#> short-read, single-end, unsorted alignment detected
#> Reading single-end BAM file
#> [0.002s]
#> Preparing lMHL report
#> [0.001s]
#> rname strand pos context coverage length lmhl
#> <fctr> <fctr> <int> <fctr> <int> <num> <num>
#> 1: chr1 + 4 CG 1 5 0.1142857
#> 2: chr1 + 7 CG 1 5 0.1142857
#> 3: chr1 + 9 CG 1 5 0.0000000
#> 4: chr1 + 12 CG 1 5 0.1142857
#> 5: chr1 + 16 CG 1 5 0.1142857
# case 3: hypermethylated read and hypomethylated read
simulateBam(output.bam.file=temp.bam, rname="chr1", XG="CT",
XM=c("h..Z..Z.Z..Z...Z.h.", "h..z..Z.z..Z...z.h."))
#> Writing sample BAM
#> [0.002s]
#> [1] 2
generateMhlReport(temp.bam)
#> Checking BAM file:
#> short-read, single-end, unsorted alignment detected
#> Reading single-end BAM file
#> [0.001s]
#> Preparing lMHL report
#> [0.001s]
#> rname strand pos context coverage length lmhl
#> <fctr> <fctr> <int> <fctr> <int> <num> <num>
#> 1: chr1 + 4 CG 2 5 0.5000000
#> 2: chr1 + 7 CG 2 5 0.5142857
#> 3: chr1 + 9 CG 2 5 0.5000000
#> 4: chr1 + 12 CG 2 5 0.5142857
#> 5: chr1 + 16 CG 2 5 0.5000000
# case 4: incompletely bisulfite-converted read and hypomethylated read
simulateBam(output.bam.file=temp.bam, rname="chr1", XG="CT",
XM=c("H..Z..Z.Z..Z...Z.h.", "h..z..Z.z..Z...z.h."))
#> Writing sample BAM
#> [0.002s]
#> [1] 2
generateMhlReport(temp.bam)
#> Checking BAM file:
#> short-read, single-end, unsorted alignment detected
#> Reading single-end BAM file
#> [0.002s]
#> Preparing lMHL report
#> [0.001s]
#> rname strand pos context coverage length lmhl
#> <fctr> <fctr> <int> <fctr> <int> <num> <num>
#> 1: chr1 + 4 CG 1 5 0.00000000
#> 2: chr1 + 7 CG 1 5 0.02857143
#> 3: chr1 + 9 CG 1 5 0.00000000
#> 4: chr1 + 12 CG 1 5 0.02857143
#> 5: chr1 + 16 CG 1 5 0.00000000
## toy examples to illustrate the logic of computations
temp.bam <- tempfile(fileext=".bam")
# case 1: fully methylated haplotype
simulateBam(output.bam.file=temp.bam, rname="chr1", XG="CT",
XM="h..Z..Z.Z..Z...Z.h.")
#> Writing sample BAM
#> [0.003s]
#> [1] 1
generateMhlReport(temp.bam)
#> Checking BAM file:
#> short-read, single-end, unsorted alignment detected
#> Reading single-end BAM file
#> [0.001s]
#> Preparing lMHL report
#> [0.001s]
#> rname strand pos context coverage length lmhl
#> <fctr> <fctr> <int> <fctr> <int> <num> <num>
#> 1: chr1 + 4 CG 1 5 1
#> 2: chr1 + 7 CG 1 5 1
#> 3: chr1 + 9 CG 1 5 1
#> 4: chr1 + 12 CG 1 5 1
#> 5: chr1 + 16 CG 1 5 1
# case 2: incompletely methylated haplotype
simulateBam(output.bam.file=temp.bam, rname="chr1", XG="CT",
XM="h..Z..Z.z..Z...Z.h.")
#> Writing sample BAM
#> [0.003s]
#> [1] 1
generateMhlReport(temp.bam)
#> Checking BAM file:
#> short-read, single-end, unsorted alignment detected
#> Reading single-end BAM file
#> [0.002s]
#> Preparing lMHL report
#> [0.001s]
#> rname strand pos context coverage length lmhl
#> <fctr> <fctr> <int> <fctr> <int> <num> <num>
#> 1: chr1 + 4 CG 1 5 0.1142857
#> 2: chr1 + 7 CG 1 5 0.1142857
#> 3: chr1 + 9 CG 1 5 0.0000000
#> 4: chr1 + 12 CG 1 5 0.1142857
#> 5: chr1 + 16 CG 1 5 0.1142857
# case 3: hypermethylated read and hypomethylated read
simulateBam(output.bam.file=temp.bam, rname="chr1", XG="CT",
XM=c("h..Z..Z.Z..Z...Z.h.", "h..z..Z.z..Z...z.h."))
#> Writing sample BAM
#> [0.002s]
#> [1] 2
generateMhlReport(temp.bam)
#> Checking BAM file:
#> short-read, single-end, unsorted alignment detected
#> Reading single-end BAM file
#> [0.001s]
#> Preparing lMHL report
#> [0.001s]
#> rname strand pos context coverage length lmhl
#> <fctr> <fctr> <int> <fctr> <int> <num> <num>
#> 1: chr1 + 4 CG 2 5 0.5000000
#> 2: chr1 + 7 CG 2 5 0.5142857
#> 3: chr1 + 9 CG 2 5 0.5000000
#> 4: chr1 + 12 CG 2 5 0.5142857
#> 5: chr1 + 16 CG 2 5 0.5000000
# case 4: incompletely bisulfite-converted read and hypomethylated read
simulateBam(output.bam.file=temp.bam, rname="chr1", XG="CT",
XM=c("H..Z..Z.Z..Z...Z.h.", "h..z..Z.z..Z...z.h."))
#> Writing sample BAM
#> [0.002s]
#> [1] 2
generateMhlReport(temp.bam)
#> Checking BAM file:
#> short-read, single-end, unsorted alignment detected
#> Reading single-end BAM file
#> [0.002s]
#> Preparing lMHL report
#> [0.001s]
#> rname strand pos context coverage length lmhl
#> <fctr> <fctr> <int> <fctr> <int> <num> <num>
#> 1: chr1 + 4 CG 1 5 0.00000000
#> 2: chr1 + 7 CG 1 5 0.02857143
#> 3: chr1 + 9 CG 1 5 0.00000000
#> 4: chr1 + 12 CG 1 5 0.02857143
#> 5: chr1 + 16 CG 1 5 0.00000000